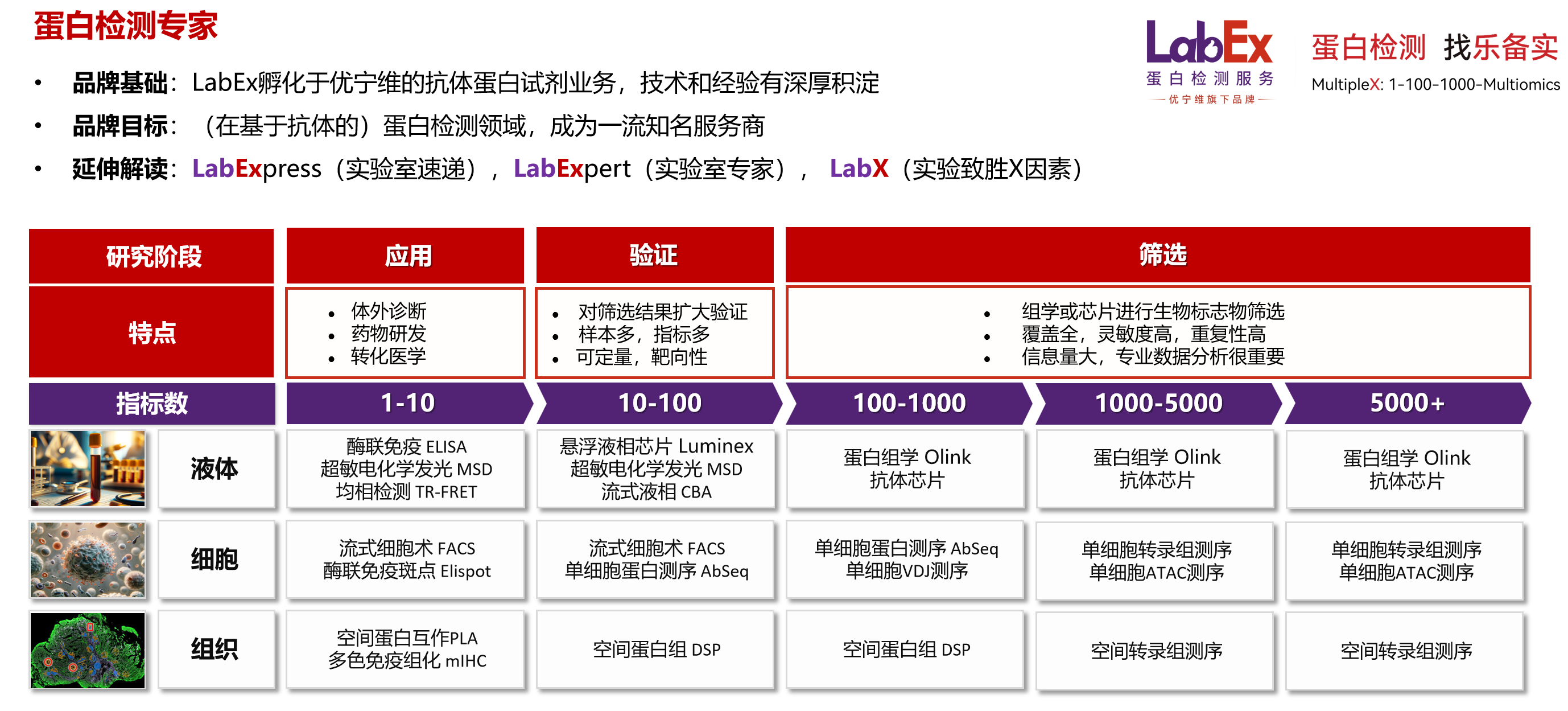
引言
T细胞终末耗竭是限制CAR-T等细胞免疫治疗长期疗效的关键因素。线粒体功能异常与T细胞耗竭密切相关,但其将代谢紊乱转化为耗竭表型的分子机制尚不清晰。
2026年3月18日,中国医学科学院血液病医院(中国医学科学院血液学研究所)王建祥教授、徐颖茜副研究员及瑞士洛桑大学路德维格癌症研究中心Ping-Chih Ho教授合作在《Nature》在线发布了题为“Proteasome-guided haem signaling axis contributes to T cell exhaustion”的研究论文。该研究发现,线粒体去极化可增强蛋白酶体活性并特异性降解线粒体蛋白,释放血红素,血红素作为转录因子调控辅因子,重塑T细胞转录调控网络。

一、研究发现
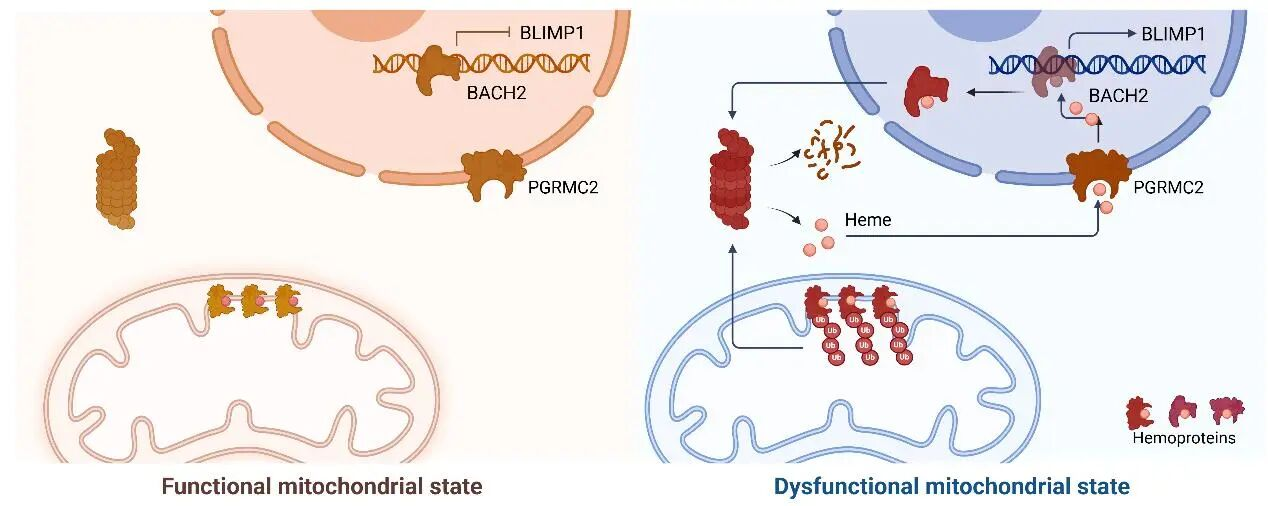
1. 线粒体去极化可显著上调T细胞蛋白酶体活性
蛋白质组学与泛素化蛋白质组学结果显示,线粒体去极化可使T细胞26S蛋白酶体亚基表达与活性明显升高,同时线粒体相关蛋白泛素化水平显著增加,且底物多为电子传递链中的血红素结合蛋白,提示蛋白酶体参与线粒体蛋白的选择性降解。
2. 蛋白酶体降解血红素蛋白释放RH,诱导T细胞耗竭
线粒体血红素蛋白经蛋白酶体降解后,细胞内调节性血红素RH水平显著上升。外源添加RH可重现T细胞耗竭表型,表现为PD-1、TIM3等抑制受体表达升高,TNF、IFN-γ等细胞因子分泌减少,增殖与杀伤功能下降,证实RH是介导T细胞耗竭的关键信号分子。
3. RH通过BACH2-BLIMP1轴重塑转录网络驱动耗竭
转录组与染色质可及性分析显示,RH可抑制干性相关转录因子BACH2的表达与功能,解除其对耗竭相关转录因子BLIMP1的抑制作用,进而上调一系列终末耗竭相关基因。突变BACH2的血红素结合位点可阻断RH诱导的耗竭表型,证实BACH2是RH发挥作用的关键靶点。
4. 蛋白酶体活性与CAR-T临床疗效呈负相关
对临床CD19 CAR-T细胞样本分析发现,患者CAR-T细胞蛋白酶体活性越高,治疗响应率越低、生存期越短,提示蛋白酶体活性可作为评估CAR-T治疗效果的潜在标志物。
5. 抑制蛋白酶体可改善CAR-T细胞功能与治疗效果
在CAR-T细胞体外制备过程中,采用低剂量蛋白酶体抑制剂硼替佐米短期处理,可降低蛋白酶体活性、减少RH生成,恢复BACH2表达并抑制BLIMP1,从而减轻CAR-T细胞耗竭,增强细胞因子分泌与肿瘤杀伤能力,并在小鼠白血病模型中显著提升治疗效果。
二、讨论
本研究阐明了线粒体去极化通过蛋白酶体-血红素-BACH2/BLIMP1信号轴驱动T细胞终末耗竭的完整机制,揭示了代谢应激向转录程序转化的关键通路。 不同于传统线粒体自噬的研究角度,本研究发现蛋白酶体介导的线粒体蛋白降解是T细胞应对线粒体损伤的重要方式,而该过程释放的血红素反而成为诱导耗竭的关键信号,拓展了代谢分子调控免疫细胞命运的认知。 在临床转化方面,蛋白酶体可作为CAR-T疗效预测标志物,而低剂量蛋白酶体抑制剂短期处理CAR-T的策略操作简便、无需基因编辑,具有良好的临床应用潜力,可为提升免疫治疗持久性提供新思路。
三、结论
线粒体去极化通过激活蛋白酶体、降解线粒体血红素蛋白释放RH,RH进一步通过BACH2-BLIMP1轴诱导CD8+T细胞发生终末耗竭;靶向抑制该信号轴可有效改善CAR-T细胞耗竭状态,增强抗肿瘤疗效,为优化肿瘤细胞免疫治疗提供了新的理论依据与干预策略。