脓毒症的高死亡率与机体抗感染免疫失调密切相关,自噬作为关键的细胞防御机制,其在脓毒症中的调控分子尚未明确。干扰素调节因子7(IRF7)在脓毒症中的作用存在争议,本研究通过体内外实验系统探究其功能及机制。结果表明,Irf7缺陷显著增加多微生物脓毒症模型小鼠死亡率,加重器官损伤并降低细菌清除效率;而IRF7过表达可逆转上述表型。机制上,IRF7作为转录因子,可直接结合Atg7、Atg9a等自噬相关基因(ATGs)启动子,同时调控自噬启动、成核、成熟等关键环节的分子表达,驱动巨噬细胞自噬体形成与自溶酶体成熟,增强胞内细菌杀伤能力。本研究明确了IRF7是脓毒症期间巨噬细胞自噬的核心调控因子,为理解脓毒症的免疫病理机制提供了新视角,也为脓毒症的靶向治疗提供了潜在分子靶点。

一、引言
脓毒症是由多微生物感染引发的全身性炎症反应,其病理过程中存在明显的免疫功能紊乱,巨噬细胞作为固有免疫的核心细胞,在细菌清除和炎症调控中发挥关键作用。自噬是细胞通过降解自身成分实现物质循环和抗感染防御的保守机制,已有研究证实自噬可保护脓毒症模型动物,但调控脓毒症相关自噬的关键分子及其作用机制仍有待阐明。 IRF7作为干扰素调节因子家族成员,在抗病毒免疫中具有明确的转录调控功能,但其在细菌感染引发的脓毒症中的作用一直存在争议。部分研究提示IRF7可能参与炎症因子调控,但缺乏对其在巨噬细胞功能及自噬调控中作用的系统研究。本研究以盲肠结扎和穿刺(CLP)诱导的多微生物脓毒症模型为研究对象,结合细胞生物学和分子生物学技术,探究IRF7对巨噬细胞自噬及细菌清除功能的调控作用,明确其在脓毒症中的分子机制。
二、材料与方法
(一)实验动物与模型构建
野生型(WT)小鼠与Irf7基因敲除(Irf7-/-)小鼠均为SPF级,采用CLP法建立多微生物脓毒症模型,通过尾静脉注射重组腺相关病毒9载体(rAAV9-Irf7)或对照载体(rAAV9-Ctrl)实现IRF7体内过表达。
(二)细胞分离与处理
分离WT小鼠和Irf7-/-小鼠的腹腔巨噬细胞及骨髓源性巨噬细胞(BMDMs),采用含10%胎牛血清的DMEM培养基常规培养。通过脂多糖(LPS)刺激模拟脓毒症炎症微环境,利用siRNA干扰技术敲低Irf7表达,质粒转染实现Irf7过表达。
(三)关键检测技术
1. 存活曲线分析:CLP术后连续7天记录小鼠存活情况。
2. 细菌清除能力检测:菌落计数法检测血液、腹腔液中细菌数量,荧光定量PCR检测细菌DNA含量。
3. 自噬相关指标检测:Western blot检测LC3、p62、ULK1、Beclin1等蛋白表达;免疫荧光染色观察自噬体形成;透射电镜分析自噬体超微结构。
4. 转录调控验证:染色质免疫沉淀测序(ChIP-Seq)分析IRF7与自噬相关基因启动子的结合情况;qPCR检测自噬相关基因(ATGs)mRNA表达水平。
5. 功能验证实验:细胞耗竭实验明确巨噬细胞、T细胞、NK细胞的作用;巨噬细胞过继转移实验验证IRF7的细胞自主性作用;自噬调节剂(Tat-Beclin1、wortmannin)干预实验明确自噬在IRF7介导的保护作用中的必要性。
三、结果
(一)IRF7是抵御多微生物脓毒症的关键分子
CLP模型中,Irf7-/-小鼠7天存活率显著低于WT小鼠,而rAAV9-Irf7介导的IRF7过表达可显著提高Irf7-/-小鼠存活率,接近WT小鼠水平(图1A)。生化指标检测显示,Irf7-/-小鼠血清髓过氧化物酶(MPO)、天冬氨酸氨基转移酶(AST)等损伤标志物水平显著升高,组织病理损伤和细胞凋亡程度加重,rAAV9-Irf7处理可逆转上述病理改变(图1B-F)。细胞因子谱分析表明,Irf7-/-小鼠晚期炎症因子(TNF-α、IL-1β、IL-6、IL-12)显著升高,IRF7过表达可抑制该过度炎症反应。
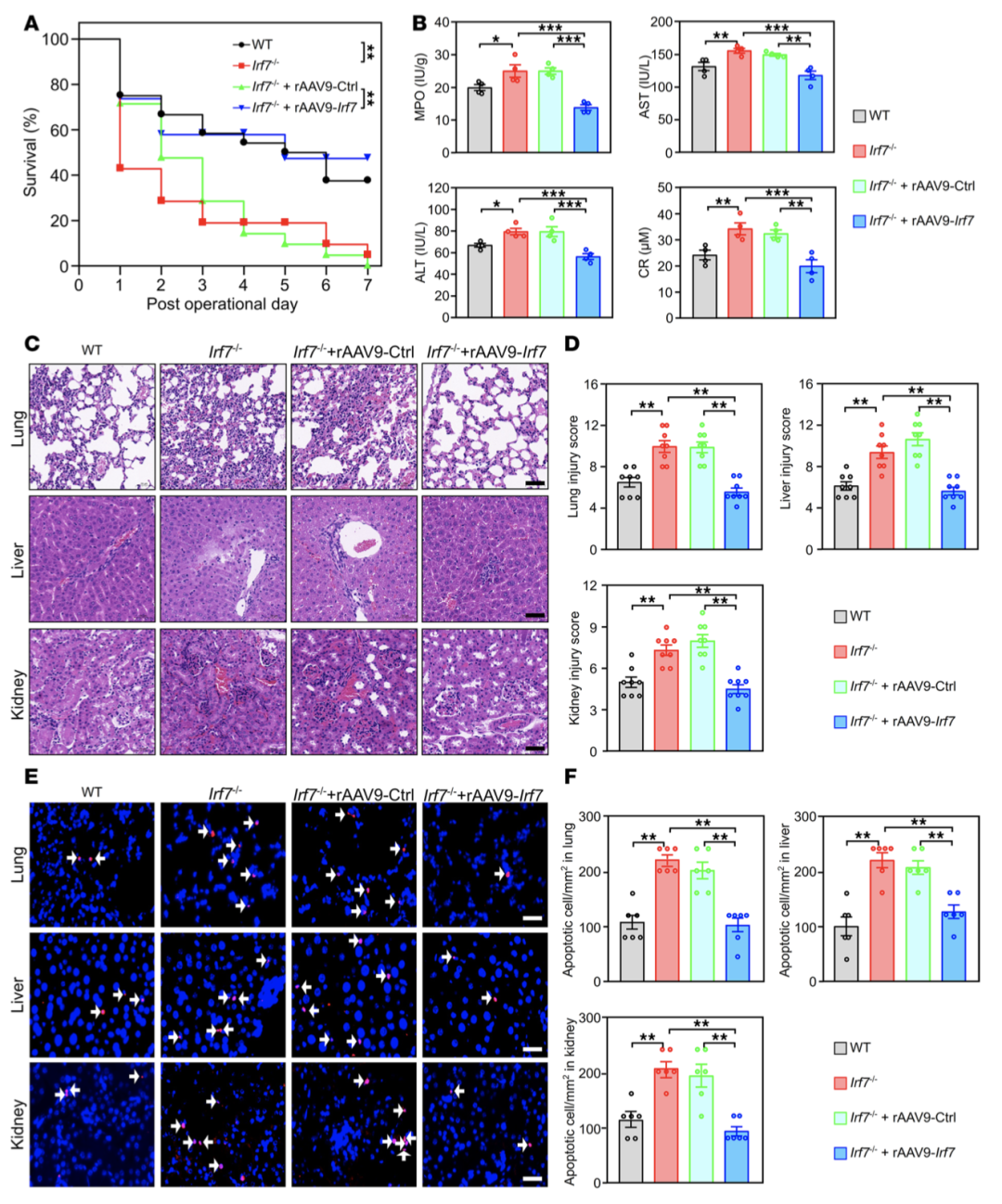
图1
(二)IRF7通过巨噬细胞发挥脓毒症保护作用
蛋白表达分析显示,CLP术后IRF7仅在巨噬细胞中特异性上调,T细胞和NK细胞中维持基线表达。细胞耗竭实验表明,巨噬细胞耗竭可显著降低WT小鼠存活率,且对Irf7-/-小鼠存活无影响,提示Irf7的保护作用依赖巨噬细胞。过继转移实验证实,WT巨噬细胞可显著提高巨噬细胞耗竭WT小鼠的存活率并减轻器官损伤,而Irf7-/-巨噬细胞无此效应(图2A-E),明确IRF7对巨噬细胞的驱动作用。
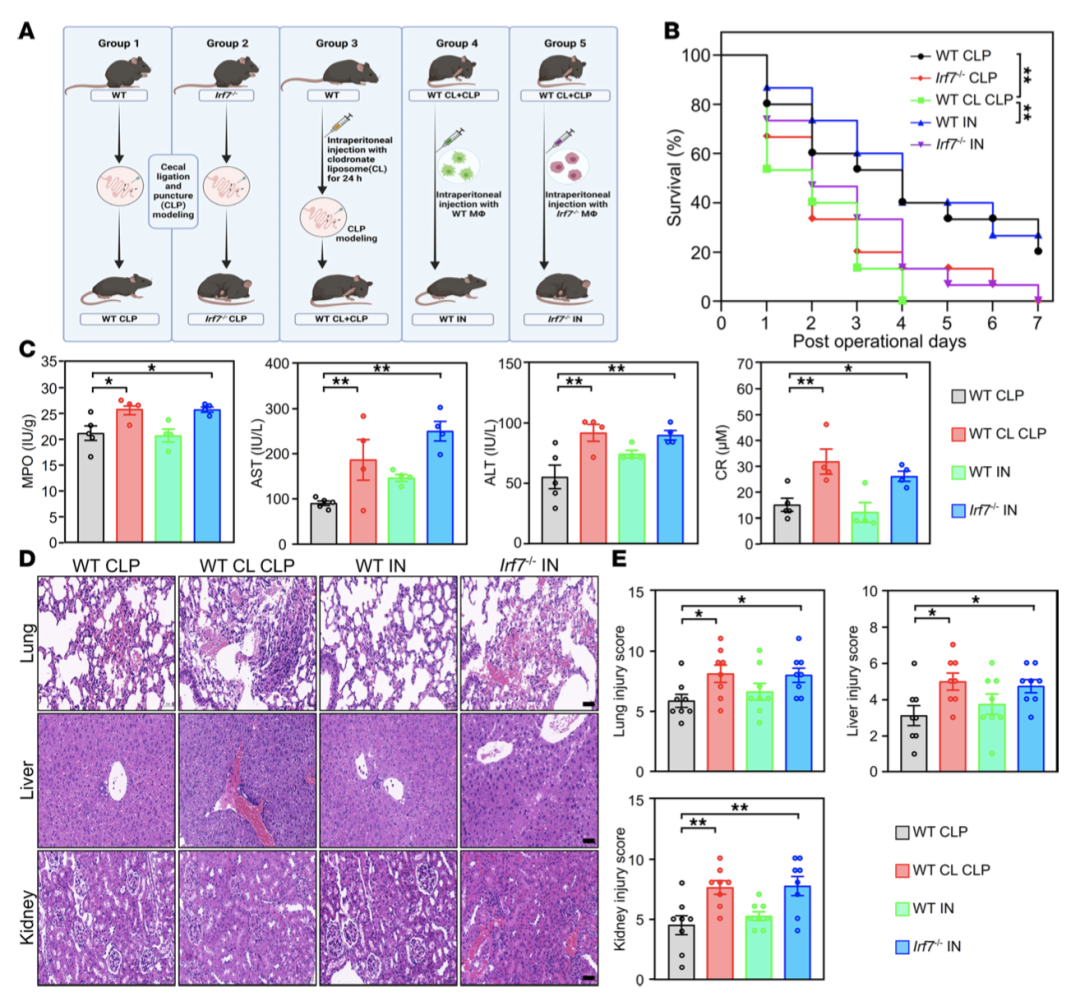
图2
(三)IRF7增强巨噬细胞的细菌杀伤能力
细菌清除实验显示,Irf7-/-小鼠血液和腹腔中细菌数量显著高于WT小鼠,rAAV9-Irf7处理可恢复细菌清除效率(图3B-C)。体外实验表明,Irf7不影响巨噬细胞吞噬能力,但可显著增强其对大肠杆菌、鼠伤寒及弧菌的胞内杀伤能力(图3D-G)。免疫荧光染色证实,Irf7-/-巨噬细胞内细菌存活数量显著增加,IRF7过表达可减少胞内细菌负荷(图3H-I)。
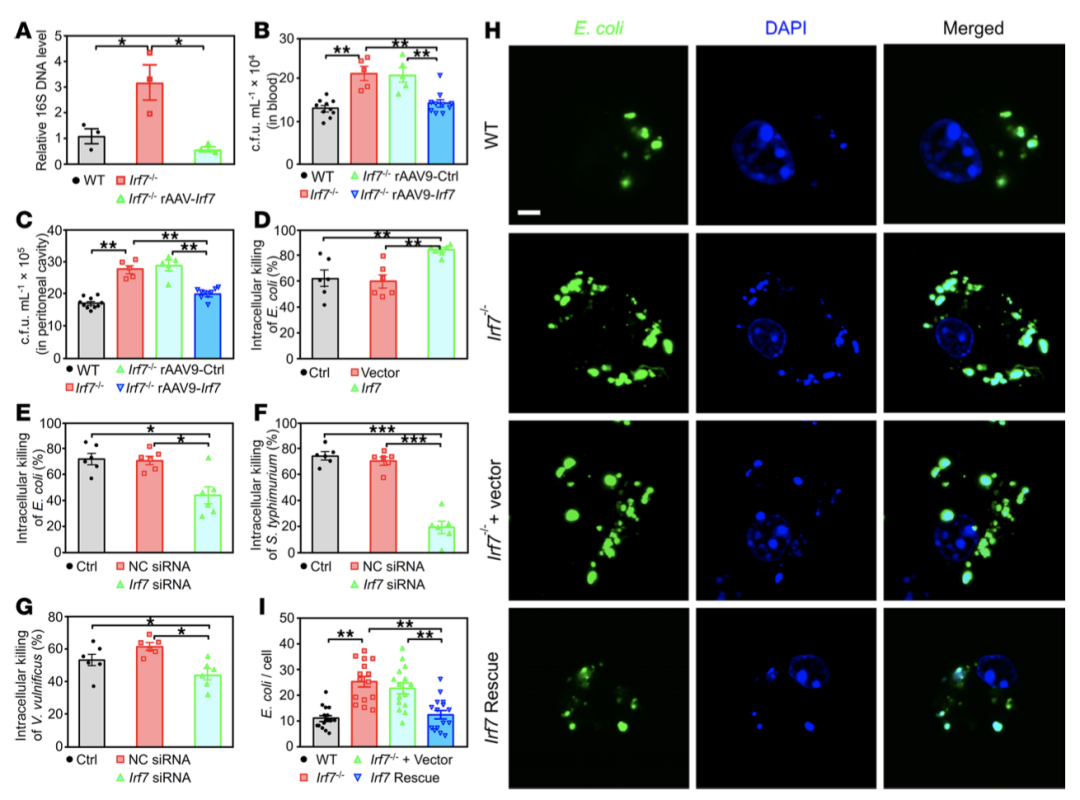
图3
(四)IRF7调控巨噬细胞自噬通路的激活
Western blot检测显示,Irf7-/-小鼠巨噬细胞中自噬标志物LC3-II表达降低,自噬底物p62蓄积;IRF7过表达可上调LPS刺激巨噬细胞中LC3-II水平,加速p62降解,并激活自噬启动(ULK1)、成核(Beclin1)、延伸(ATG7)、成熟融合(RAB7、LAMP2)及线粒体自噬(DRP1)相关分子(图4A-C)。超微结构和免疫荧光分析证实,IRF7缺失显著减少自噬体数量,IRF7拯救可恢复自噬体形成(图4D-E)。
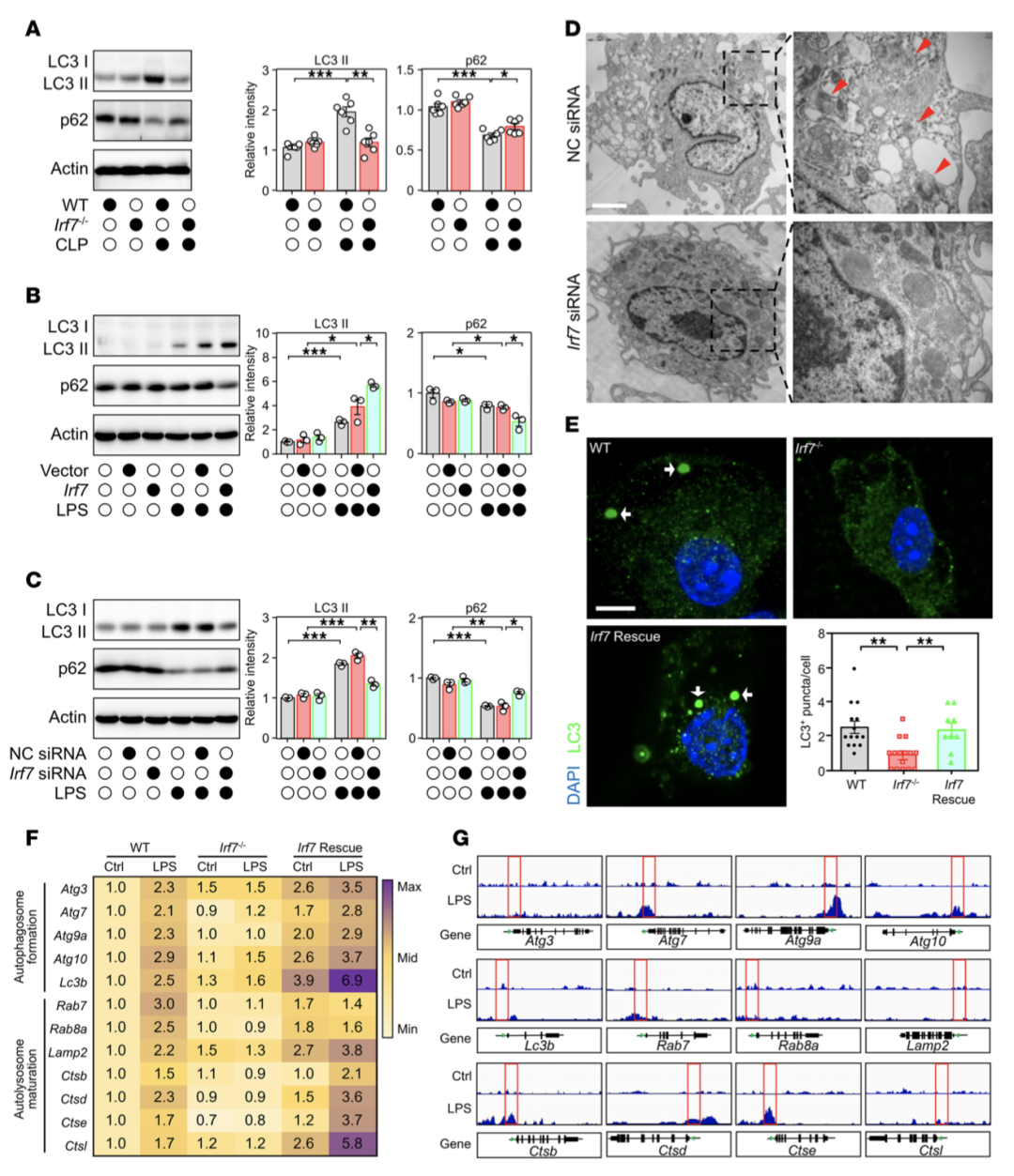
图4
(五)IRF7通过转录调控自噬相关基因表达
ChIP-Seq结果显示,IRF7可直接结合Atg7、Atg9a、Atg10、Rab7等7个ATGs的启动子区域,同时调控Atg3、Lc3b等其他ATGs的表达(图4F-G)。KEGG通路富集分析表明,这些靶基因主要参与内吞、溶酶体处理及自噬等细菌清除相关通路,证实IRF7是脓毒症诱导自噬的主转录调控因子。
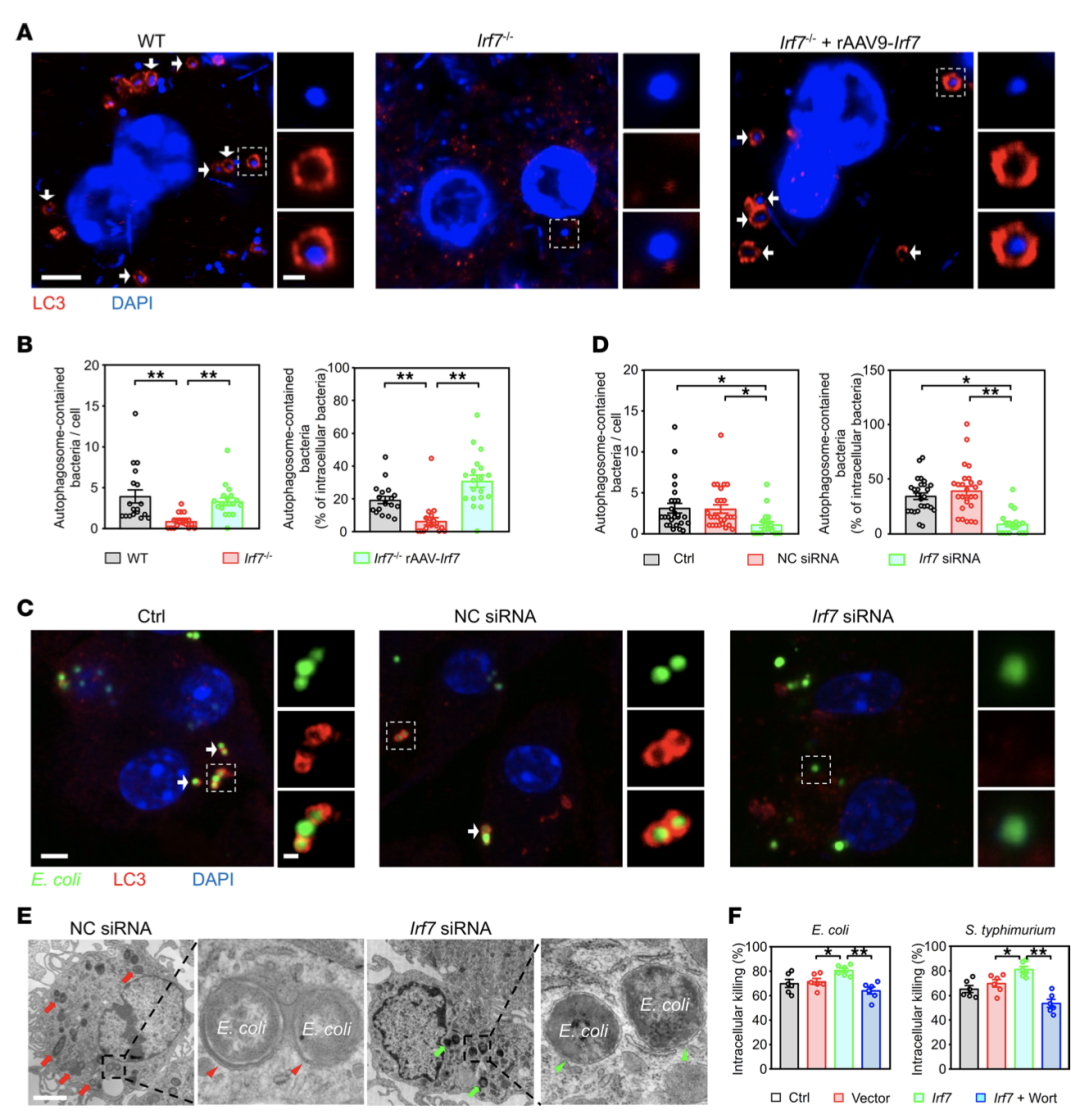
图5
(六)自噬介导IRF7的细菌杀伤及脓毒症保护作用
自噬调节剂干预实验显示,自噬诱导剂Tat-Beclin1可增强巨噬细胞细菌杀伤能力,而自噬抑制剂wortmannin可阻断IRF7介导的细菌清除效应(图5F)。体内实验中,Tat-Beclin1可提高Irf7-/-小鼠存活率,wortmannin则削弱rAAV9-Irf7的保护作用(图6A-D),证实IRF7通过自噬通路发挥脓毒症保护作用。
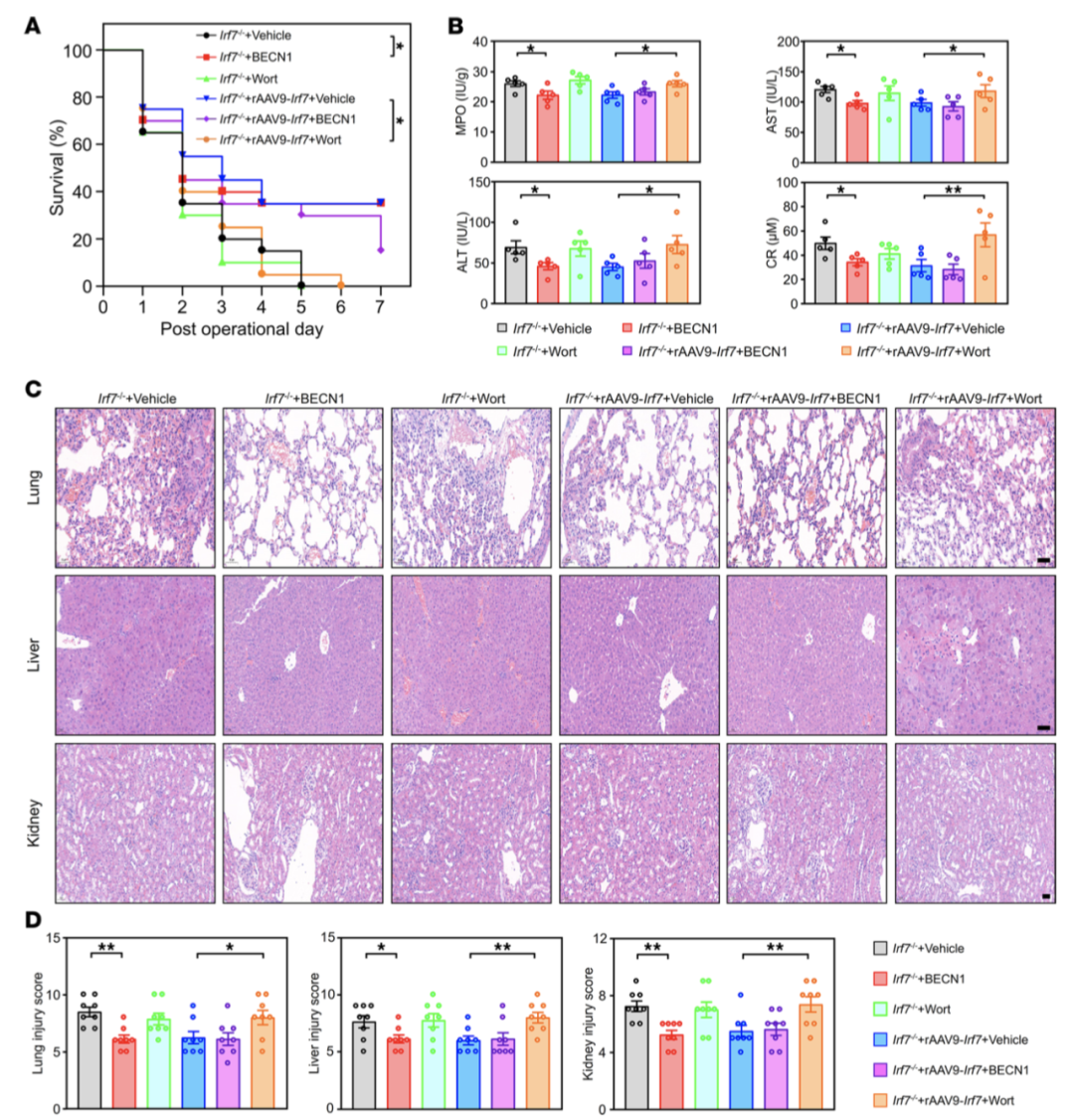
图6
四、结论
本研究明确了 IRF7 在脓毒症中的关键保护作用,其核心机制在于通过转录调控巨噬细胞自噬通路增强细菌清除能力。此前 IRF7 的研究多集中于抗病毒免疫,本研究首次揭示其在细菌感染引发的脓毒症中的功能,成功解决了自噬在脓毒症中调控因子不明的关键科学问题。作为转录因子,IRF7 通过直接结合 Atg7、Atg9a 等自噬相关基因(ATGs)启动子,同时间接调控下游分子表达,全面激活自噬启动、成核、延伸及成熟融合等关键环节,这一调控模式为理解自噬网络的复杂性提供了新的分子机制。研究进一步证实 IRF7 对巨噬细胞的调控具有特异性,其保护作用不依赖 T 细胞和 NK 细胞,明确了巨噬细胞在 IRF7 介导的脓毒症防御中的核心地位。
五、LabEx蛋白检测服务